补体过度活化在阿尔茨海默病(AD)和额颞叶痴呆(FTD)等神经系统疾病中介导了小胶质介导的突触消除,但是大脑中如何调节补体活性仍然大多未知。
约翰·霍普金斯大学医学院的研究者们发现分泌型神经元蛋白Nptx2与补体C1q结合,从而在大脑中调节其活性。Nptx2缺乏的小鼠显示补体活性增加,依赖C1q的小胶质突触吞噬,以及兴奋性突触的丧失。在神经炎症培养模型和老化TauP301S小鼠中,通过腺相关病毒(AAV)介导的神经元Nptx2过度表达足以抑制补体活性并改善小胶质介导的突触丧失。并证明Nptx2调节大脑中的补体活性和小胶质突触消除,而Nptx2浓度的减少可能会加重FTD患者中补体介导的神经退行性。研究成果以“The neuronal pentraxin Nptx2 regulates complement activity and restrains microglia-mediated synapse loss in neurodegeneration” 为题,发表在Science Translational Medicine(IF:17.1)上。

研究结果
1. NPTXs在体外结合C1q并抑制经典补体途径(CCP)的活性
研究者发现C1q与外周蛋白pentraxins有高亲和性结合。通过实验,确定C1q与神经元蛋白NPTXs相互作用,并形成复合物。实验证实C1q能够与NPTXs的pen-traxin结构域结合。进一步的研究表明,NPTXs可以调节补体介导的细胞溶解和细菌杀伤作用,通过抑制C1q活性来阻止补体的功能。总的来说,这些结果揭示了NPTXs在调控补体活性方面的重要作用,可能对神经退行性疾病的治疗具有潜在的意义。

2. Nptx2缺失会增加CCP活性,并导致体内兴奋性突触的丢失
在阿尔茨海默病(AD)中,Nptx2在死后大脑和脑脊液中浓度降低,其浓度与疾病状态、认知表现和病情进展相关。在精神分裂症患者的脑脊液中,Nptx2的浓度也降低,Nptx2缺乏的小鼠表现出与精神分裂症相关的行为缺陷。
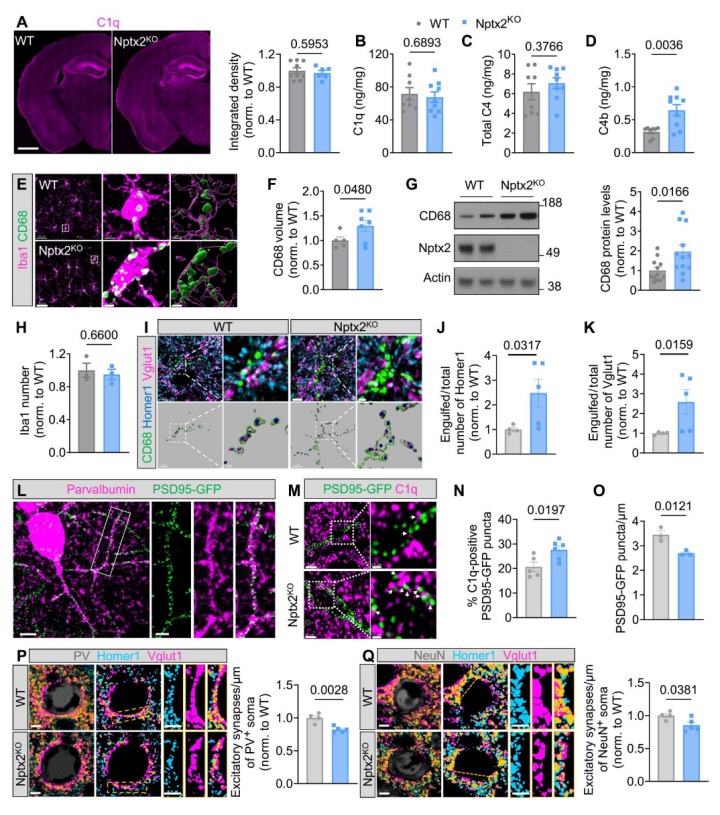
鉴于Nptx2作为补体调节因子的发现,研究者进一步探查了Nptx2KO小鼠大脑中是否存在失调的补体活性。首先,对2至3个月大的Nptx2KO小鼠大脑进行了C1q的免疫染色,发现C1q免疫荧光强度在Nptx2KO小鼠和野生型(WT)小鼠的大脑中相似,这一结果在C1q酶联免疫吸附法(ELISA)中得到了验证。虽然Nptx2KO大脑中下游C4的全长蛋白浓度也未改变,但激活的C4(C4b)的含量显著增加,与Nptx2缺乏的大脑中补体活性增加一致。
小胶质细胞是补体介导的突触修剪的执行者。鉴于补体活性的增加,研究者想知道小胶质细胞是否在Nptx2KO大脑中表现出吞噬性表型。通过免疫组化染色和免疫印迹法,发现Nptx2KO小鼠皮层中的吞噬体小胶质细胞标记CD68的免疫反应性明显增加,但小胶质细胞数量在Nptx2KO大脑皮层中没有改变。研究者进一步证实Nptx2主要在兴奋性神经元中表达,并且在兴奋性突触标记物蛋白Vglut1和突触素附近检测到Nptx2的突触标记。因此,推测Nptx2的丧失可能使突触更容易受到补体介导的小胶质细胞吞噬。通过共免疫染色观察兴奋性突触后标记物Homer1和突触前标记物Vglut1与小胶质细胞CD68+溶酶体的关系,发现在Nptx2KO大脑皮层中,Homer1和Vglut1免疫反应性在CD68+溶酶体内明显增加,这表明小胶质细胞对兴奋性突触的吞噬在Nptx2KO大脑中增加。
这些结果表明,补体介导的修剪导致Nptx2KO皮层中的兴奋性突触丧失,而抑制性突触在Nptx2缺乏的条件下基本不受影响。
3. 补体抑制可以挽救Nptx2KO大脑中的突触损失
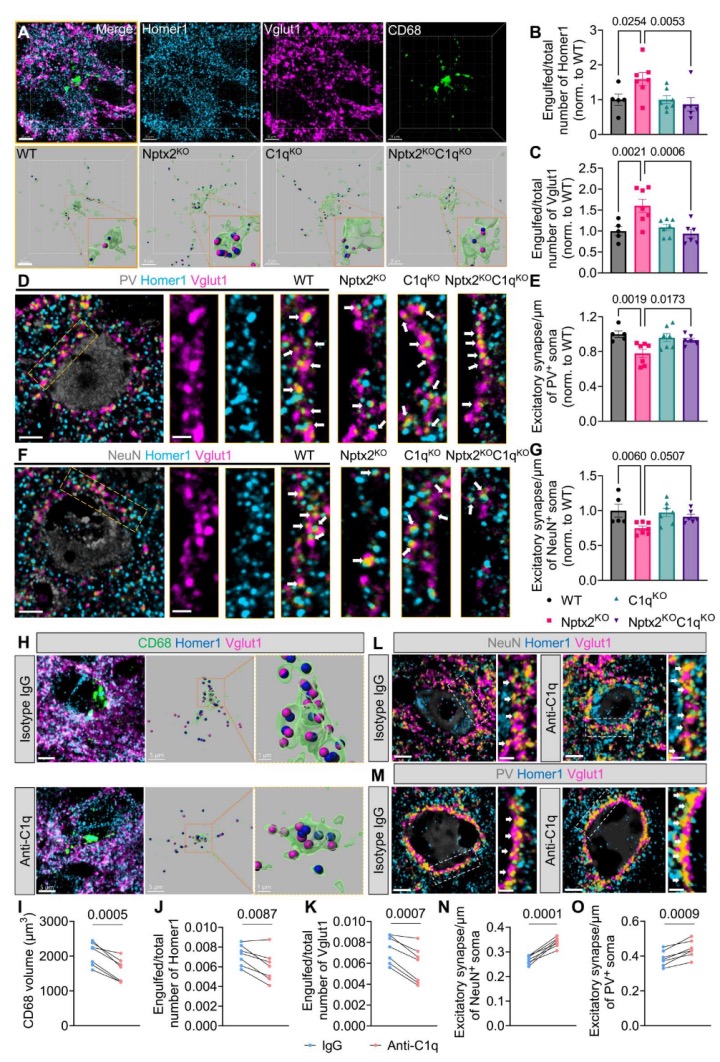
为了测试Nptx2KO大脑中的突触丧失是否依赖于补体系统,研究人员比较了缺乏C1q、Nptx2或两者的小鼠(Nptx2KO;C1qKO双缺失小鼠)。研究发现,在Nptx2KO大脑中,小胶质细胞CD68+溶酶体体积增加,表明小胶质细胞活性增强。然而,在缺乏C1q的情况下,CD68+溶酶体体积并未增加,这表明这种小胶质细胞现象与C1q有关。
进一步实验发现,在Nptx2KO大脑中,小胶质细胞对兴奋性突触的吞噬显著增加,而缺乏C1q时,这种增加的吞噬现象得到减轻。这暗示了C1q在Nptx2KO大脑中调控小胶质细胞对兴奋性突触的吞噬过程。
为了验证这一现象,研究人员还注射了能有效抑制C1q活性的抗体到Nptx2KO小鼠的大脑中。结果显示,C1q阻断抗体的注射导致小胶质细胞对兴奋性突触的吞噬减少,并且突触密度得到恢复。这表明补体系统在Nptx2缺失导致的突触丧失过程中起着重要作用。
最后,研究人员使用C1q阻断抗体直接注射到大脑中,进一步证明了C1q在突触丧失过程中的作用。这项实验显示,抑制C1q活性可减少小胶质细胞对兴奋性突触的吞噬,并恢复突触密度。
4. 在神经炎症神经元-小胶质细胞共培养模型中,神经元Nptx2过表达限制了补体介导的神经毒性
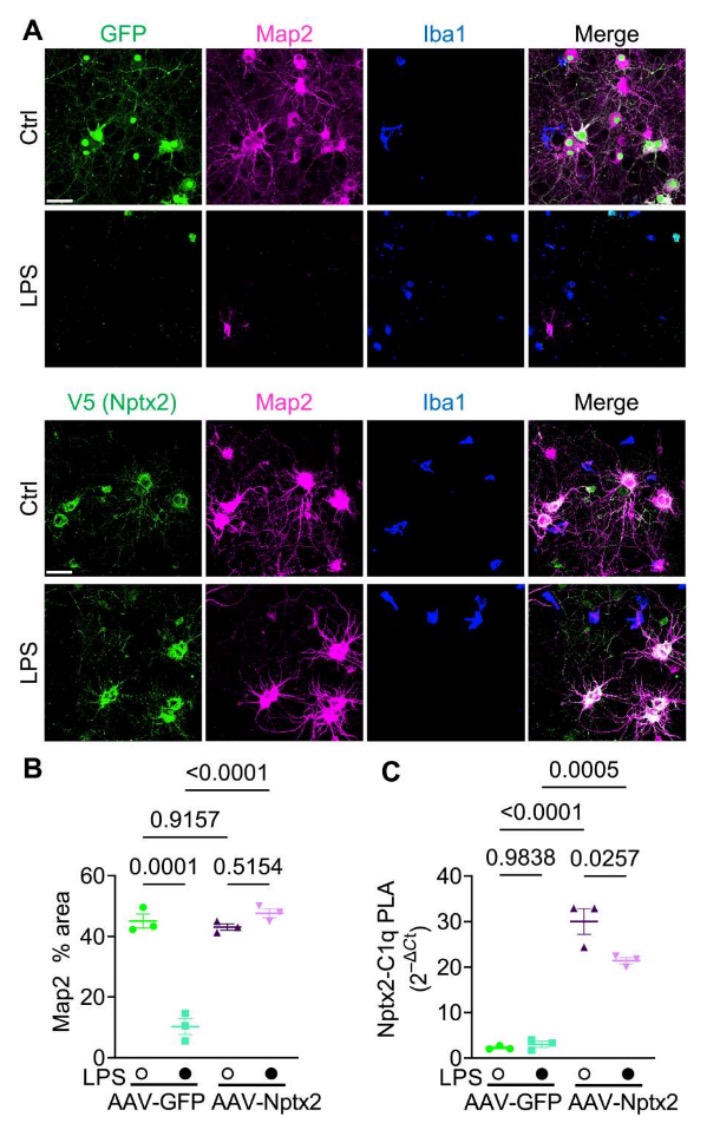
为了进一步验证Nptx2是否可以改善神经毒性,研究人员在将小胶质细胞加入神经元培养之前,使用携带V5标签的Nptx2或GFP的AAV病毒对神经元培养进行感染,让神经元过表达Nptx2。结果显示,与过表达GFP的对照组相比,过表达Nptx2的神经元可以预防由LPS活化小胶质细胞导致的神经元丧失。研究人员推测,分泌的Nptx2能够结合并阻止细胞外的C1q,从而抑制补体系统的激活。为了验证这一假设,建立了一个称为近距离连接测定(PLA)的实验方法。结果显示,通过AAV介导的Nptx2过表达或添加rNptx2,显著增加了共培养培养基中C1q-Nptx2复合物的丰度。
5. AAV诱导的Nptx2过度表达可以改善P301S小鼠的突触损失
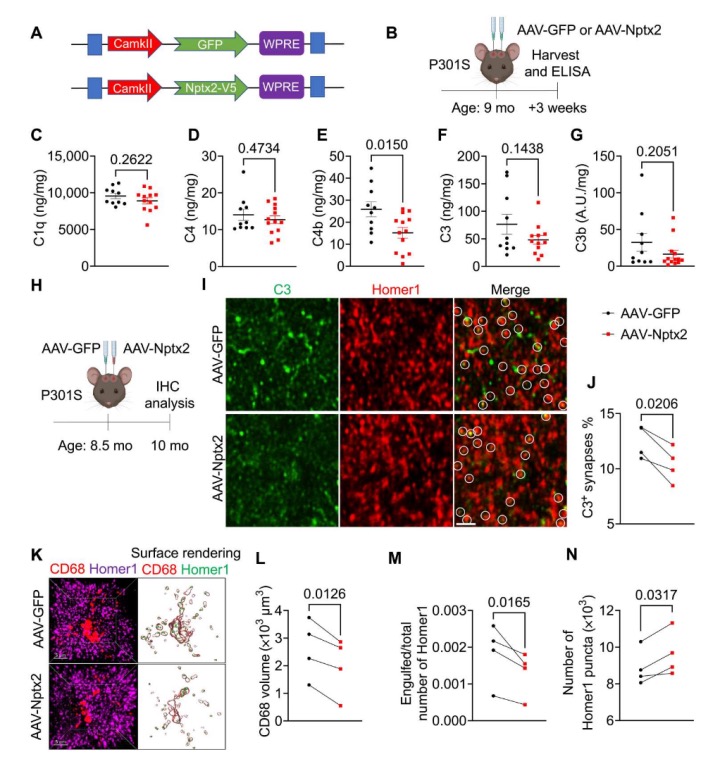
为了验证Nptx2是否可以调节补体系统并改善突触损失,研究团队注入了过表达V5标记的Nptx2或GFP的AAV病毒到P301S小鼠的海马中。结果显示,Nptx2过表达后,补体系统中的C4b显著降低,而总C3和C3b的浓度也有所减少。此外,Nptx2过表达导致小胶质细胞中补体蛋白的活性显著降低,并减少了突触的丢失。这进一步证明了Nptx2过表达足以限制补体系统的过度激活并改善突触损失。
在进一步实验中,研究团队将AAV-Nptx2注射到8.5个月龄的P301S小鼠的海马,并在注射后6周通过免疫组织化学分析了大脑。结果显示,Nptx2过表达不影响Tau蛋白的磷酸化程度和胶质细胞的活化。然而,Nptx2过表达显著减少了小胶质细胞中的补体蛋白结构,并降低了兴奋性突触的小胶质细胞吞噬。这与突触密度的增加相一致,进一步证明Nptx2过表达可以改善补体介导的小胶质细胞突触修剪。
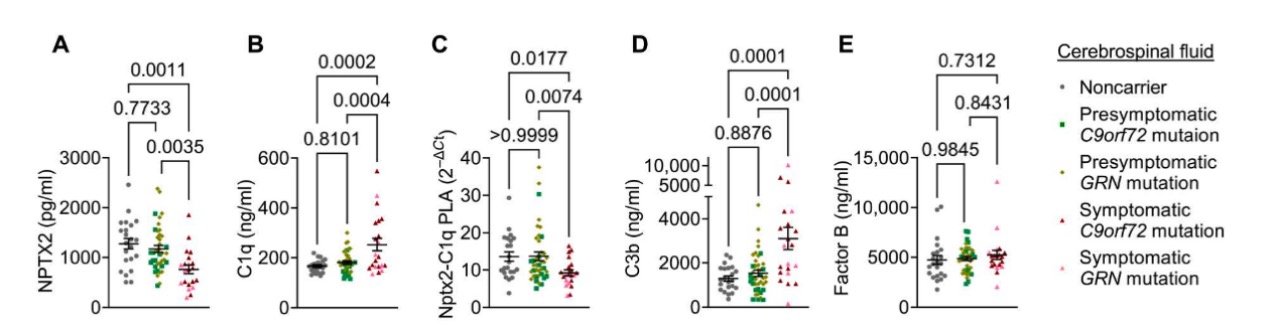
6. Nptx2、C1q和Nptx2-C1q复合浓度在遗传FTD CSF中发生变化
补体在神经退行性疾病中被认为是导致突触损失和神经元损害的主要驱动因素。先前的研究表明,在AD患者的脑脊液中,C3和C4的总量以及通过CCP激活引发的C3和C4的裂解产物增加。进一步的研究发现,症状患者的CSF中C1q浓度明显增加,而Nptx2-C1q复合物的浓度降低。实验结果显示在CSF中存在着Nptx2和C1q的复合物,且在症状患者中的含量较低。此外,CSF中的C3b浓度也增加,支Nptx2作为调节CCP活性的潜在因子。
研究总结
这篇文章的主要内容是关于神经元蛋Nptx2在调节补体活性和限制小胶质细胞介导的突触丧失方面的作用。研究发现,在神经退行性疾病中,补体活性被认为是导致突触丢失和神经元损害的主要驱动因素。
在研究中,通过使用细胞培养和动物模型,研究者发现Nptx2能够与补体蛋白C1q结合并阻止其激活,从而抑制补体系统对神经元的损伤作用。此外,研究还表明AAV助力Nptx2的过表达可以减轻补体引起的突触丧失,并降低小胶质细胞对神经元的摧毁。此外,研究还发现在遗传性神经退行性疾病患者中,Nptx2的浓度降低,而C1q的浓度增加,导致补体活性增强。这一现象可能导致补体系统的失衡,从而加剧神经退行性疾病的发展。
总的来说,这项研究揭示了Nptx2在调节补体活性和抑制小胶质细胞介导的突触丧失中的关键作用,为神经退行性疾病的治疗提供了新的治疗靶点。同时,该研究还强调了补体系统在神经退行性疾病中的重要作用,为深入理解这些疾病的发病机制提供了重要线索

公众号底部菜单栏【新功能】上线!
病毒实验帮
免费在线学习《国自然热点研究》、《数据库及软件操作教程》
一键下载《病毒使用手册》、《高分文献》
还有不定时的送新书、抽奖活动,赶紧来扫码关注一波吧